Epidemiology, Population Health Genetics and Phenotypic Diversity of Sickle Cell Disease in India
R Balgir
Citation
R Balgir. Epidemiology, Population Health Genetics and Phenotypic Diversity of Sickle Cell Disease in India. The Internet Journal of Biological Anthropology. 2007 Volume 1 Number 2.
Abstract
The sickle cell disease is characterized by crescent-moon-shaped red blood cells, resulting in hand and foot syndrome, chronic anemia, jaundice, serious frequent infections, painful episodes, vaso-occlusive crisis, enlarged spleen, retardation of development and growth, and damage to vital body organs, barring acute chest syndrome, leg ulcer, lung blockage and priapism complications in India. The disease is inherited as an autosomal intermediate dominant fashion. The sickle cell patients in India do not show severe clinical manifestations unlike the African patients due to interaction of α-thalassemia with sickle cell disease, high fetal hemoglobin level, and maintenance of life at low hemoglobin level. Kulozik and coworkers have described independent Asian origin of sickle cell haplotype based on the presence in India and East Saudi Arabia, but recent findings from India question this independent and unicentric origin of tribal population in India. In this paper, epidemiology, population genetics and phenotypic diversity of sickle cell disease have been discussed in the light of recent findings in India.
Notes
This was an invited paper presented in Indo-US Symposium on “Genetic Disorders: Focus on Hemoglobinopathies” held during 29-31 October 2006, Banaras Hindu University, Varanasi, Uttar Pradesh, India
This paper is dedicated to my wife, Mrs. Baljeet Kaur Balgir, for her unstinted support, pains taking care, and cheerfully looking after me through all adversities of life.
Introduction
India is a great and vast country with diverse and multiple castes, ethnic groups, religions, occupations, economic strata, languages, socio-cultural traditions, genetic heritages and life-styles and practices. Moreover, this country has absorbed in so many racial, religious, socio-cultural, linguistic and genetic elements that have given rise to amalgamation and conglomeration of all the constituent features, representing the cohesive unity in diversity in the true sense.
This paper critically discusses the epidemiology, population health genetics and phenotypic diversity of sickle cell disease in the light of recent findings in India.
Clinical Profile of Sickle Cell Disease
The sickle cell disease is an inherited hematological disorder characterized by banana, crescent-moon or sickle-shaped human red blood cells. This abnormality results in painful episodes, chronic anemia, epistaxis, enlarged spleen, serious frequent infections, and damage to vital body organs 1 , 2 , 3 . Under normal circumstances, red blood cells are flexible and round, and move easily through blood vessels to carry oxygen to all parts of the body for about 90-120 days. Sickled cells have relatively small oxygen contact area and increased blood viscosity, and impede normal circulation in small blood vessels, resulting in ischemia and infarction. In people with sickle cell anemia, these irregular-shaped red cells become rigid and sticky, and die prematurely, resulting in a chronic shortage in the body. While traveling through small blood vessels, they can get stuck and slow down or block blood flow and oxygen to certain parts of the body. This blockage produces pain and serious complications. These effects can, however, vary from person to person depending on the type of sickle cell disease, nutritional status, and other concomitant factors. Signs and symptoms of the sickle cell disease include:
-
Anemia. Elongated sickle cells are fragile. They break apart easily and die, leaving chronical shortage of red blood cells to carry oxygen to tissues — a condition known as anemia. Without enough red blood cells in circulation, body can't get the oxygen to energize the tissues. That's why anemia causes fatigue.
-
Episodes of pain. Periodic episodes of pain, called crises, are a major symptom of sickle cell anemia. Pain develops when sickle-shaped red blood cells block blood flow through tiny blood vessels to chest, abdomen and joints. Pain can also occur in bones, varying in intensity and last for a few hours to a few weeks. Some people experience only a few episodes of pain, whereas, others experience a dozen or more crises in a year. If a crisis is severe enough, painkillers are to be injected intravenously, and the patient may need hospitalization.
-
Hand-foot syndrome. Swollen hands and feet are often the first signs of sickle cell anemia in babies. The swelling is caused by sickle-shaped red blood cells blocking blood flow out of the hands and feet, accompanied by pain and fever.
-
Jaundice. Jaundice is a yellowing of the skin and eyes that occurs because of liver damage or dysfunction. Occasionally, people who have sickle cell anemia have some degree of jaundice because the liver, which filters harmful substances from the blood, is overwhelmed by the rapid breakdown of red blood cells. In people with dark skin, jaundice is visible as the eye retina becomes yellowish.
-
Frequent infections. Sickle cells damage spleen, an organ that fights infections, making it more vulnerable to infections. Doctors commonly prescribe antibiotics to infants and children to prevent potential life-threatening infections, such as pneumonia.
-
Stunted growth. Red blood cells provide oxygen and nutrients needed for the growth. A shortage of healthy red blood cells can slow down growth in infants and children and delay puberty in teenagers.
-
Vision problems. Some people with sickle cell anemia experience vision problems. Tiny blood vessels that feed eyes become plugged with sickle cells. This damages the retina — the portion of each eye that processes visual images.
-
Vaso-occlusive crisis is caused by sickled red blood cells that obstruct capillaries and restrict bloodflow to an organ, resulting in ischemia, pain, and organ damage. Because of its narrow vessels and function in clearing defective red blood cells, the spleen is frequently affected. It is usually infarcted before the end of childhood in individuals suffering from sickle-cell anemia. This auto-splenectomy increases the risk of infection from encapsulated organisms; preventive antibiotics and vaccinations are recommended for those with such asplenia. Liver failure may also occur with time. Bone is also a common target of vasoocclusive damage, especially when the bone is particularly weight-bearing. Such damage may result in avascular necrosis (especially of the femur) and bone deterioration. The pain experienced by sickle-cell patients is also due to the bone ischemia.
-
Aplastic crisis. An acute worsening of the patient's baseline anemia producing pallor, tachycardia, and fatigue. Reticulocyte counts drop dramatically during the illness and the rapid turnover of red cells leads to the drop in hemoglobin (Table-1). Most patients can be managed supportively; some need blood transfusion.
Although some generalization about the genotype-phenotype correlation may be made, sickle cell patients sharing identical genotypes exhibit considerable heterogeneity in clinical symptoms in India 1 , 2 , 4 . Some of the common symptoms seen in patients suffering from sickle cell anemia include:
-
Anemia
-
Intermittent jaundice
-
Severe joint pains
-
Recurrent infections
The following complications are either absent or rare in the Indian patients 4 :
-
Acute Chest Syndrome
-
Leg Ulcer
-
Lung blockage
-
Painful erections in men (priapism)
Genesis of Hemoglobin and its Structure
At the time of conception, a person receives one set of genes from the mother (egg) and a corresponding set of genes from the father (sperm). The genes exist on structures inside cells called chromosomes. The combined effects of many genes determine some traits (hair color and height, for instance). Similarly, the other gene pairs determine the other characteristics. People have twenty-two identical chromosome pairs and the twenty-third pair is either XX or XY chromosome that determines the sex of a person. One of each pair is inherited from the father, and one from the mother. One of the genes may be altered by any of a variety of “accidents” that can occur in nature. This altering event, called mutation, occurs extremely rarely.
The deoxyribonucleic acid (DNA) molecule is the fundamental genetic material that determines the arrangement of the amino acid building blocks in all proteins. Segments of DNA that code for particular proteins are called genes. The genes that encode α-globin chains are located on chromosome 16 and the non-α-globin chains are on 11 th chromosome 5 . The gene that controls the production of β-globin subunit of hemoglobin (Hb) is located on one (chromosome 11) of the 46 human chromosomes. Hemoglobin is a protein made up of four chains called globins and four iron (heme) groups. With the exception of the very first weeks of embryogenesis, one of the globin chains is always alpha (α). A number of variables influence the nature of the non-α chain in the hemoglobin molecule. The fetus has a distinct non-α chain called gamma (λ). After birth, a different non-α globin chain, called beta (β), pairs with the α-chain. The combination of two α-chains and two non-α chains produces a complete hemoglobin molecule (a total of four chains per molecule). Complex biophysical characteristics of the hemoglobin tetramer permit the exquisite control of oxygen uptake in the lungs and release in the tissues that is necessary to sustain life.
The combination of two α-chains and two λ-chains form “fetal” hemoglobin, termed “hemoglobin F”. Hemoglobin F is a poor participant in hemoglobin S fibril formation and thus inhibits sickling. With the exception of the first 10 to 12 weeks after conception, fetal hemoglobin is the primary hemoglobin in the developing fetus. The combination of two α-chains and two β-chains form adult (A) hemoglobin, also called “hemoglobin A”. Although hemoglobin A is called “adult”, it becomes the predominant hemoglobin within about 18 to 24 weeks of birth 6 . The genes involved in sickle cell disease control the production of a protein in red cells called hemoglobin. Hemoglobin binds oxygen in the lungs and delivers it to the peripheral tissues, such as the liver and muscles. Most of the people have two normal genes for hemoglobin. Sometimes, one parent is carrier of S and other is carrier of β-thalassemia or other structurally abnormal hemoglobin like D or E, then that condition is known as double heterozygosity 7 . This may lead to Hb S β-thalassemia, Hb SD or Hb SE disease.
The sickle cell disease is inherited as autosomal recessive manner. If one of the parents is normal (Hb AA) and the other has sickle cell trait (Hb AS), there is 50% chance of each pregnancy for having a sickle cell trait child, but none of the children will have sickle cell disease (Hb SS). If both the parents have sickle cell trait (Hb AS), then there is 25% chance of each pregnancy for normal child (Hb AA), 50% chance of each pregnancy for sickle cell trait (Hb AS) child, and 25% chance of each pregnancy for sickle cell disease (Hb SS) child (Fig.-I). If one parent is sickle cell disease (Hb SS) and the other a sickle cell trait (Hb AS), then 50% chance of each pregnancy to be sickle cell disease (Hb SS) and 50% chance of a child with sickle cell trait (Hb AS).
Brief History of Sickle Cell Disease
James B. Herrick, a Chicago physician, gave the first description of sickle cell disease, who noted that a student patient from Grenada (West Indies) had an anemia characterized by unusual “elongated and sickle-shaped” red blood cells 8 . However, Mason 9 used the term sickle cell anemia for the first time.
Using the new technique of protein electrophoresis, Pauling et al. showed that the hemoglobin from patients with sickle cell disease is different than that of normals 10 . This made the sickle cell disease as the first disorder in which an abnormality was known to be at fault in a protein.
In 1957, Ingram sequenced sickle hemoglobin and showed that a glutamic acid at position 6 was replaced by a valine in sickle cell disease. Using the known information about amino acids and the codons, they were able to predict the mutation in sickle cell disease 11 . This made possibility of cure of sickle cell disease, the first genetic disorder whose molecular basis was known at that time.
Allison in 1954 reported a relationship between sickle cell trait and Plasmodium falciparum malaria that the sickle cell gene is maintained as true polymorphism in the population by having a selective survival advantage to the heterozygotes 12 .
In 1984, bone marrow transplantation (BMT) in a child with sickle cell disease produced the first reported cure of the disease. The transplantation was done to treat acute leukemia. The child's sickle cell disease was cured as a side-event. The procedure nonetheless set the precedence for later transplantation efforts directed specifically at sickle cell disease. Three large clinical trials--one in Belgium, another in France and a third in the US--have reported results of BMT for 116 patients with sickle cell disease. All but three of the patients received marrow from a matched related donor--a relative whose marrow type was similar to the patient's. The remaining three were transplanted with cord blood, which like bone marrow is rich in stem cell--the cells that produce most of the body's blood cells. Of the 116 patients, 91 (78 percent) were alive and disease-free two to four years following BMT. An additional 16 were alive, but had rejected the donor marrow or cord blood (graft rejection), and continued to have symptoms of sickle cell disease.
In a multicentric study of Hydroxyurea in Sickle Cell Anemia, hydroxyurea became the first (and only) drug proven to prevent complications of sickle cell disease by enhancing the production of fetal hemoglobin in 1995. However, the detrimental side effects of urea, subsequently, did not allow the continuation use of this drug.
Pre-implantation genetic diagnosis (PGD) could provide couples with another way to avoid passing on sickle cell disease. A couple who was a carrier of sickle cell disease had twin babies who were completely free of the sickle cell gene with the application of a technique called pre-implantation genetic diagnosis (PGD), in which the fertilized embryos were tested for the disorder before implantation in the woman's uterus 13 . Further refinement in this technology could become a powerful diagnostic tool in the assistive reproductive technology (ART) in the near future.
Population Genetics and Sickle Cell Phenotypic Diversity
The sickle cell disease was unknown to the present day Indian people before the independent discovery in 1952. The first cases of sickle hemoglobin in India were reported by Dunlop and Mazumder 14 who detected five cases of sickle cell trait and three presumptive cases of sickle cell anemia among the tea garden labourers of Upper Assam, originating from the tribal populations of Bihar and Orissa states. In the same year, Lehmann and Cutbush 15 reported the presence of sickle cell trait among the aboriginal (Pre-Dravidian) tribe (Toda) of the Nilgiri Hills in Southern India. In fact, for the first time, the sickling was detected in India by PK Sukumaran who was working at Cancer Research Institute (CRI) laboratory in Bombay at that time under Prof. L.D. Sanghvi, before Lehmann and Cutbush (1952) were approached by CRI for confirmation for the existence of sickle cell gene in India (Personal discussion with PK Sukumaran in 1986). Subsequently, there have been several surveys in the Nilgiri Hills and other areas by different foreigner and Indian researchers. Mention may be made among others of Negi 16 and Basu 17 who worked among the tribals of Bastar in Madhya Pradesh.
The sickle cell hemoglobinopathy has remained a neglected field of research in India and the magnitude of the problem has never been properly appreciated by our physicians, hematologists, pediatricians, geneticists and anthropologists, in spite of the fact that sickled red blood cells (RBCs) were detected in the blood of patients originating from Orissa in Tea Garden labour population in Assam as early as 1952. This was largely because of the fact that most of the subsequent reports spread a misconception that the sickle cell gene was confined only to Tribal People or some Scheduled castes in India 18 . The other reason was the hereditary nature and no cure for the disease. However, the first review on sickle cell hemoglobin in India was done by Balgir and Sharma 19 , and followed by Balgir 2 , 20 , 21 , 22 , which highlighted the wide spread distribution of the sickle cell disorders in India.
The distribution of sickle cell disorders in various communities of different states of India shows the prevalence range and also the name of community with the highest incidence (Fig.-II). The highest prevalence has been recorded in the state of Orissa (1-44.4%), followed by Madhya Pradesh (1-40.0%; including Chhattisgarh), Tamil Nadu (1-40.0%), Andhra Pradesh (1-35.7%), Assam (1-35.5%), Maharashtra (0.8-35.0%), Gujarat (1-31.4%), Kerala (1-30.0%), Uttar Pradesh (1.5-18.5%), Karnataka (1-8.0%), Rajasthan (1-5.7%), West Bengal (1-1.7%), and Bihar (0.8%; including Jharkhand) in the decreasing order 19 , 20 , 22 . The sickle cell disorder is common mostly in scheduled tribes, infiltrated in scheduled castes in most of these states except in Orissa, where the general castes have shown the highest prevalence unlike in other states 20 , 22 , 23 , 24 , 25 , 26 .
Studies in the state of Orissa based on hospital as well as random population surveys have revealed interesting story quite different from rest of India. The sickle cell gene is highly prevalent among the general castes (0.3-20.7%), scheduled castes (0-8.9%) and scheduled tribals (0-5.5%) in Orissa 24 , 25 . The question arises, why the sickle cell gene is so common among the general castes of Orissa as compared to the other states of India? The general castes, scheduled castes and scheduled tribes in Orissa do not live in biosocial isolation and the biological contact between them is possible. Moreover, the answer is provided by the fact that the general castes of other states have not yet been so extensively investigated as that of the state of Orissa. There is also likelihood that probably the general caste groups of Orissa have the socio-cultural, ethnic, genetic and economic status at par with the scheduled castes and scheduled tribes of the other states of India 18 . Further, which seems to be the most plausible reason, is the practice of marital consanguinity amongst some of the castes, namely, Agharia, Kulita, Khandayat, Teli, Gaud, etc. who prefer and practice parallel and cross-cousin marriages 27 . This leads to intense inbreeding and multiplication of the abnormal sickle cell gene more rapidly in the population. Comparatively, the smaller size of the effective breeding population is also another reason for the high occurrence of this congenital abnormality.
Prospective studies carried out in Orissa have revealed statistically significant differences in hematological indices between sickle cell trait and sickle cell disease, sickle cell-β ++ thalassemia, and sickle cell-β + -thalassemia; between sickle cell disease and sickle cell-β ++ thalassemia, and sickle cell-β + -thalassemia; and between sickle cell-β ++ thalassemia and sickle cell-β + -thalassemia were observed with variable symptomatology and clinical manifestations. Genetic heterogeneity of sickle cell-β-thalassemia is noticed in India 28 , 29 . Both mild (African) and severe (Mediterranean) forms of sickle cell-β-thalassemia (i.e., β ++ thalassemia and β + -thalassemia or β o -thalassemia) are encountered, suggesting the gene flow from the East Mediterranean, Asian Gulf, sub-Sahara and East Africa reflecting historical events and gene migrations in the region, having implications in community health genetics in India 29 .
Genetic diversity and heterogeneity with respect to the genetic markers indicated not only inter-tribal admixture but also the diffusion with other racial groups of India. This may be due to continuous pressures of gene diffusion from different waves of people penetrating their territory and habitats in Orissa in the course of invasions and migrations from the North-Western (Indo-Aryan) and North-Eastern (Mongoloid) parts of India during the ancient and historical times 24 , 25 .
Evolutionary Trend for Inverse Relationship
The sickle cell disease is an autosomal recessive disorder that causes anemia, joint pain, a swollen spleen, and frequent severe infections. If natural selection eliminates individuals with detrimental phenotypes from a population, then why do harmful mutant alleles persist in a gene pool? A disease can remain prevalent when heterozygotes have some other advantage over individuals who have two copies of the harmful alleles. When carriers have advantages, because of balanced polymorphism, then that advantages allow a detrimental allele to persist in a population. This form of polymorphism often entails heterozygosity for an inherited illness that protects against an infectious illness. It illustrates balanced polymorphism because carriers are resistant to malaria, an infection by the parasite
For the first time, studies have shown an evolutionary trend for inverse relationship between the sickle cell allele and G-6-PD deficiency, and sickle cell and β-thalassemia allele in a cross-section of malaria endemic
Origin and Diversity of Sickle Cell Haplotypes
“ No one knows whence and at whose call
came pouring endless inundations of men
rushing madly along -- to loose themselves in the sea:
Aryans and non-Aryans, Dravidians and Chinese
Scythians, Huns, Pathans and Moghuls --
all are mixed, merged and lost in one body.
Now the door has opened to the West
and gifts in hand they beckon and they come --
they will give and take, meet and bring together,
none shall be turned away
from the shore of this vast sea of humanity
that is India.”
--- Rabindranath Tagore
Human invasions or migrations from different corners of the country are well documented in the Indian history, which have resulted in human proliferation with varied genetic elements. India is a country with vast reservoir of abnormal hemoglobins and thalassemias. Most of the abnormal hemoglobins like Hb C, D, E, H, J, K, L, M, Q, S, Chandigarh, Koya Dora, Lepore, Lucknow, Norfolk, Rambha and hereditary persistence of fetal hemoglobin were either first found in India or in the individuals of Indian origin abroad 6 . A large number of hemoglobin variants so identified in India testify the human genetic diversity in India. Human migrations with the fall of Indus Valley Civilization and scatter in different parts of India are virtually the most valuable information 30 , 31 . It is interesting to highlight the spread of various human hemoglobin variants as a result of these invasions, migrations, and trade connections and/or due to slavery, with special reference to sickle cell gene in India.
In the African continent, Livingstone 32 gave a hypothesis of a single sickle cell mutation in the Mediterranean region and, subsequently, the diffusion of the same occurred to other neighboring countries during Muslim expansions. Contrary to the above hypothesis, the β S -haplotypes linked with Eastern Saudi Arabia are found absent in the Indian Muslim populations. Moreover, the high prevalence of sickle cell gene among the tribals of India cannot be explained on the basis of above postulations 2 .
Nagel 33 and Pagnier et al 34 have described three independent mutations, namely Senegal, Benin, Bantu and later on Cameroon in African continent itself. They have also traced different routes of human migrations in the African continent based on these independent sickle cell mutations.
Kulozik et al 35 have described independent Asian origin of sickle cell haplotype based on the presence of this haplotype in India and East Saudi Arabia. This sickle cell haplotype is associated with high level of fetal hemoglobin; interacting with α-thalassemia and mild clinical manifestations of the sickle cell disease in India. The spread of this haplotype was accounted for the Muslim expansions during 1st or 2nd Century AD in India. Further, 50% of the β S -haplotypes found in the unrelated cases from Pune (India) as reported by Kulozik et al 35 were different from the β S -haplotypes of East Saudi Arabia and India.
Labie et al 36 while confirming the previous independent origin of sickle cell haplotype in India have proposed the unicentric origin of the tribes in India and further postulated the possibility of spread of Indus valley civilization, which was probably harboring the sickle cell gene.
This study has serious kinds of lacunae:
-
This hypothesis goes against the earlier
35 viewpoint in the sense that Muslim expansions in India probably brought the sickle cell gene from Saudi Arabia during 1st or second century A.D. -
Sickle cell has not been observed in Muslim populations in India
-
Indication of prevalence of sickle cell gene in India (during Indus Valley Civilization) event before the Muslim expansions took place in India
30 ,31 . High frequency of this mutation in the nontribals does not justify the unicentric origin of the sickle cell mutations in India2 ,25 . Tribal diversity in India does not warrant the unicentric origin of the tribes in India2 ,22 .
It has been reported that the sickle cell disease is 30% prevalent in North Kerala especially in the hilly tracts of Wynaad district 37 . These patients attend to Medical College, Calicut. A significant point is that these patients have relatively low levels of fetal hemoglobin 38 , contrary to Kulozik et al 35 findings.
The Cameroon β S -haplotype was reported by Gupta et al 39 in Gond tribe of Central India (Madhya Pradesh) and in Mala scheduled caste in Andhra Pradesh 40 . Majumder et al 41 have shown the presence of Benin and Bantu β S -haplotype in Agharia and Gaud (general caste) communities from Orissa, which further supports rather testify the presence of Negroid elements in the Indian populations (Fig.-III).
The origin and dispersal of β S -mutation is still a controversial issue in the world including in India. Different views have been expressed by different investigators, varying from a single independent mutation 32 to multiple mutations 33 , 34 at several occasions, from a single geographical location to multicentric origin (s), from diffusion to a large scale migrations and altogether independent β S -mutation in the world. Several β S -mutations namely, Senegal, Benin, Bantu, Cameroon and Arab-Indian have been identified. The later β S -mutation has been encountered in East Saudi Arabian peninsula and India. Our recent studies in India question the independent Asian origin of β S -mutation and also of the unicentric origin of the tribal populations in India 2 , 42 . Inadequate population based studies in India present with inconclusive inference and controversies. However, these findings have special significance from anthropological and evolution point of view for the diversity of mankind in India.
Studies based on restricted fragment length polymorphism (RFLP) in the β-globin gene cluster in Orissa are the most fascinating 41 . We could analyse haplotype most frequently associated with the sickle cell allele, i.e. -++ haplotype. This haplotype is a part of the Arab-Indian haplotype. However, the atypical haplotype -+- (part of Benin haplotype) was also detected in significant frequency in Agharia and Gaud populations of Orissa 41 . The haplotype --- (part of the Bantu haplotype) was detected among the Gauds. These Benin and Bantu haplotypes which are exclusively found in the African Continent, but prevalent in the Indian populations especially in Orissa (Table-2) testified the presence of Negroid elements in India and their migrations from Africa in the distant past 2 , 41 , 42 .
To conclude, it is likely that some of the sickle cell carriers, which the Muslim invaders took with them as slaves from India or Indian traders or Indian Gypsies 43 made slaves in Saudi Arabia might have, subsequently, multiplied there during the course of history, which shows β S -haplotypes identical with those of Indian populations 42 .
Epidemiology of Sickle Cell Disease
About 250 million people, i.e. 4.5% of the world population are carriers and about 3,00,000 infants are born with a major hemoglobinopathies every year 44 . The high frequency of sickle cell gene in certain areas leads to a high birth rate of homozygotes.
As a result, sickle cell disorders account for about 70% of the worldwide hemoglobin disorders 44 . As per WHO report 45 , 60 million carriers of sickle cell and 1,20,000 sickle cell homozygotes are added every year in the world. With a population of 1000 million at the new millennium (2000) year and a birth rate of 25 per 1000 liveborns, there would be about 45 million carriers and about 15,000 infants born each year with hemoglobinopathies in India 6 . However, the exact share of sickle cell disease is still unknown in India. There were 1,86,096 sickle cell anemia cases present in the Indian subcontinent according to one estimate 46 . Based on 1981 census figures of population in India, it was estimated that there were 24,34,170 carriers and 1,21,375 sickle cell homozygotes among the tribes in India 47 . Based on the prevalence rates of sickle cell hemoglobin, it was estimated that there were over 50,00,000 persons carriers and two lakhs homozygous sickle cell disease cases among tribals alone in India 48 .
However, our estimates indicate that there were 3-4 million people suffering from sickle cell hemoglobinopathy in Orissa alone according to census data of 1981 and one-fourth of a million constituted the scheduled tribes in Orissa 49 . We contended that these figures are still under estimates and the actual number is much higher in India. An alarming numbers of them are added every year in India. It has been estimated that on an average there are 19.32% people who are victims of hemoglobinopathies in Orissa or every fifth person in Orissa has this condition 50 , 51 . Of these 19.32% of the people, 13.2% suffer from sickle cell disorders (sickle cell trait = 8%; sickle cell disease = 4.0%; sickle cell-β-thalassemia = 1.2%) or every 8 th person is afflicted with this disorder in Orissa 51 .
The combined average allele frequency of the three most predominant abnormal hemoglobins, i.e. hemoglobin D (Hb D; 0.86%), hemoglobin E (Hb E; 10.9% in North Eastern India) and sickle cell (Hb S; 4.3%) has been found to be 5.35% in India 21 .
Abnormal hemoglobins affect human physiology, growth and development, and sexual maturation; and subsequently, the fertility and mortality. The body mass index of patients with sickle cell disease was found very low (16.9±2.9) as compared to trait (19.4±2.8) or control (19.7±2.8) cases 52 . Mukherjee and Gangakhedkar 53 have made similar observations for the sickle cell children in Mumbai. The persons afflicted with sickle cell are generally mild to moderately undernourished, susceptible to diseases especially to infectious diseases, which lead to high morbidity and mortality 52 . The onset of age at menarche is delayed for 1-3 years in homozygous sickle cell afflicted girls, depending upon the nutritional status, parasitic infestations, and other afflicted conditions 54 . Homozygous females have reduced fertility, but pregnancy occurs (Table-3) and is associated with high degree of maternal morbidity, fetal wastage and neonatal mortality 55 . High fertility has been observed among the heterozygote carriers of sickle cell disease 55 . Infant mortality is very high among the affected families of sickle cell disease.
The pattern of death in persons who have sickle cell anemia is bimodal, with the first peak occurring in childhood and the 2 nd occurring in people in their late 30s. Deaths during childhood are related to infectious causes, whereas those during adulthood are due to organ failure from repeated tissue destruction 2 , 18 , 56 .
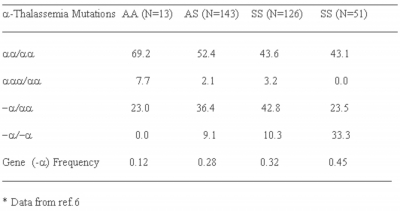
It is interesting to note that the sickle cell patients in India do not show severe clinical manifestations unlike the African patients and can survive upto 3rd or 4 th decade of life 18 , 56 . For this, interaction of α-thalassemia with sickle cell disease (Table-4), high fetal hemoglobin level (Table-1) and maintenance of life at low level of hemoglobin, etc. is responsible 57 . However, some sickle cell trait cases those are otherwise just like normals, but show severe clinical manifestations and are blood transfusion dependant. This indicates heterogeneity of sickle cell disease in India.
Epidemiological studies of sickle cell disorders showed age specific mortality and relatively lower fitness of sickle cell homozygotes. The homozygotes of sickle cell disease die earlier (younger) than the heterozygotes (trait or carriers). It was interesting that as we move from lower to higher age categories, the number of cases of sickle cell hemoglobinopathy goes on decreasing under natural environmental conditions showing probably age specific mortality (Fig.-IV). The average life span of sickle cell homozygotes is about 20 years as against the normal life span of 50 years of heterozygotes 2 , 18 , 56 .
It is surprising that the infant/neonatal mortality rate is almost three times higher in sickle cell carrier parents (80.3) than in the normal (26.7) parents per 1000 livebirths 48 , 49 . The neonatal mortality per mother is four times higher in sickle cell carrier couples than in the normal couples. These findings are detrimental to the progeny of sickle cell trait couples and are substantially contributing to the high neonatal/infant mortality rate in India 51 . For the first time, our studies have revealed that hereditary causes, apart from nongenetic factors, are also responsible for the high neonatal/infant mortality in India 51 . Infant Mortality Rate of 83 deaths for every 1000 births in 2003 was dubiously the highest in Orissa with a target to reduce it to 60 by the year 2005, comparable with an Indian (National) average of 60 in the year 2003. However, it (IMR) remained 83 even in the year 2005, but reduced to 73 in the year 2007.
High degree of marital consanguinity, caste/class and geographical endogamy, lack of medical facilities, psycho-social prejudices, certain irrational traditions and beliefs aggravating the nutritional status and health, heavy cost of treatment, poor economy, backwardness in all spheres of life, etc. are some of the salient features of these vulnerable people in India. However, a joint venture of antenatal care and inductive screening seems to be the most fruitful strategy for hemoglobinopathies in India. An Integrated National Preventive and Management Health Policy for Hemoglobinopathies in India is repeatedly over emphasized 7 . Unless locality based and need specific health care approach is evolved which would be appropriate, acceptable, accessible, and affordable, the target of health for all will remain a Utopian dream in India.
Opening of antenatal and prenatal diagnostic facilities near the vicinity of at risk populations, providing genetic/marriage counseling, imparting of health education, making people economically viable by different means of employment, etc. are the basic approaches which may be able to alleviate the sufferings of these down-trodden masses in India.
Figure 1

Figure 2

Figure 3

Figure 4

Figure 5

Figure 6

Figure 7

Figure 8
Conclusions
The sickle cell disease is characterized by banana or sickle-shaped red blood cells; and abnormality results in recurrent fever, chronic anemia, epistaxis, jaundice, high level of fetal hemoglobin, painful episodes, enlarged spleen, serious frequent infections, and damage to vital body organs. The complications of acute chest syndrome, leg ulcer, lung blockage and priapism are absent in the Indian patients.
The sickle cell disease is an inherited hematological disorder with core prevalence in Central India. The exact share of sickle cell disease is still unknown in India. The frequency varies from population to population (tribals as well as nontribals) depending upon the practice of community and territorial endogamy between 1-44.4%, but commuted average sickle cell disorder frequency of 4.3% has been worked out in India. An alarming number of the cases are added every year in India. The compound heterozygosity is equally common in India.
Infant/neonatal mortality and reproductive wastage is very high for sickle cell disease in India. Pattern of death in persons suffering from sickle cell anemia is bimodal, with the first peak occurring in childhood and the 2 nd in their late 30s.
The origin and dispersal of β S -haplotype is still a controversial issue varying from single to multiple mutations, and from diffusion to large-scale migrations, from single geographical location to multicentric origins, and altogether independent mutations in the world.
The recent findings of Indus valley civilization (site) indicate the prevalence of hereditary anemia (sickle cell disease or β-thalassemia) in the Indian subcontinent about 2000-5000 BC.
An evolutionary trend for inverse relationship between sickle cell allele and G-6-PD deficiency and β-thalassemia alleles has been observed with replacement for β-thalassemia and G-6-PD deficiency alleles against the sickle cell allele in India.
An Integrated National Preventive and Management Health Policy for Hemoglobinopathies in India is repeatedly over emphasized.
Acknowledgements
Author is grateful to Prof. N. K.Ganguly, Director General, Indian Council of Medical Research, New Delhi for providing the necessary facilities and to all those authors whose work has been quoted in this article.