Osteogenesis Imperfecta In A Set Of Nigerian Twins – A Case Report
I Fajolu, V Ezeaka, O Elumelu, O Onabajo, C Ananti, E Iroha, M Egri-Okwaji
Keywords
nigeria, osteogenesis imperfecta, twins, type ii
Citation
I Fajolu, V Ezeaka, O Elumelu, O Onabajo, C Ananti, E Iroha, M Egri-Okwaji. Osteogenesis Imperfecta In A Set Of Nigerian Twins – A Case Report. The Internet Journal of Pediatrics and Neonatology. 2012 Volume 14 Number 1.
Abstract
Osteogenesis imperfecta is a generalized disorder of connective tissue especially the bones and is the commonest cause of osteoporosis and lethal short-limbed dwarfism. We report a case of type II osteogenesis imperfecta in a set of Nigerian monochorionic twins.The twins were female and delivered by emergency caesarean section at term, Twin 1 had a length of 46cm (both below 3rd percentile), blue sclera, frog leg like posture, with widened anterior and posterior fontanelles with metopic and sagittal sutural diastases. There were multiple abnormal angulations and tender swellings of the upper and lower limbs, a short ribcage with Pectus excavatum and was dyspnoeic. Skeletal survey showed multiple healed fractures, with callus formation affecting all long bones with fresh unhealed fractures of the left humerus, radius, ulna & right tibia and fibula in the first twin and fractures of the left tibia in the second twin. There was no family history of similar condition and there was no consanguinity. The babies were managed conservatively and discharged for follow up.This is to remind physicians that though Ostegenesis imperfecta can be autosomally and recessively inherited, spontaneous mutations can also occur and that the more lethal types II and III may be commoner in Nigeria.
Introduction
Osteogenesis imperfecta (OI) is an inherited disorder of connective tissue especially the bones and is the commonest cause of osteoporosis and the leading cause of lethal short-limbed dwarfism and crippling skeletal dysplasia. People with OI have either a qualitative or quantitative defect in type I collagen resulting in fragile bones, fractures with minimal or no trauma and sometimes low bone mass. The classical manifestation is a triad of fragile bones, blue sclerae, and early deafness. It has also been described as Brittle bone syndrome, Adair-Dighton syndrome, Van der Hoeve's syndrome and Ekman-Lobstein syndrome.
There are no racial, ethnic or sex differences and age of onset varies with type. The condition is caused by mutations in the genes that code for type I collagen1 and mutations in one of the two genes - COL1A1 and COL1A2 - are found in 80% to 90% of people with OI.2 Type I collagen fibers are synthesized as procollagen molecules and are found in the bones, organ capsules, fascia, cornea, sclera, tendons, meninges and dermis. Structural defects are either point mutation (80%) or single exon splicing defects (20%). Quantitative defects with null mutations lead to reduced amount of normal collagen.
The incidence of osteogenesis imperfecta is 1 in 20,000- 50,000 live births.3 It is classically inherited as an autosomal dominant trait however a recessive pattern of inheritance may result from mutations in other genes e.g. CRTAP (Cartilage Associated Protein) or LEPRE1 (Leucine Proline Enriched Proteoglycan1) genes4,5.
Sporadic germ-cell mosaicism due to new mutations in the COL1A1 or COL1A2 gene may explain cases occurring in families with healthy parents that have more than one child with the disorder.
The original Sillence classification of OI in four types (OI type I - IV), was based on clinical findings and mode of inheritance with a radiological sub-classification of type II in A, B, C, 6,7however this has been expanded over the years to include OI types I-VIII.4,8
Types V, VI, VII
The objective of this report is to highlight the occurrence of this rare condition in a set of Nigerian twins and also emphasize the possibility of a higher incidence of the more lethal forms in Nigeria.
Case Report
A set of twins (both female) who were delivered at 39 weeks gestation via elective caesarean section on account of poor obstetric history (two previous still births) to a 29 year old para 3 + 0 mother. The antenatal period was essentially uneventful. There was no history of consanguinity or malformations in the family however the cause of death in the still births was not known as a post mortem was not done and a description of the babies could not be obtained. The first child is an eight year old female who is alive and well with no abnormalities
Twin 1
The Apgar scores were 5 and 8 in the first and fifth minutes respectively, the birth weight was 2300g and the length was 46cm (both below 3 rd percentile). Baby had blue sclera, frog leg like posture, widened anterior and posterior fontanelles with metopic and sagittal sutural diastases. There were multiple abnormal angulations and tender swellings of the upper and lower limbs, a short ribcage with Pectus excavatum and she was dyspnoeic.
A diagnosis of a term low birth weight small for gestational age female neonate with osteogenesis imperfecta was made. A skeletal survey that was done revealed multiple healing fractures, with callus formation affecting all long bones with new fractures of the left humerus, radius, ulna & right tibia and fibula. The serum calcium, phosphorus and alkaline phosphatase were within normal limits.
Twin 2
She had APGAR scores of 8 in first minute and 9 in the fifth minute, her birth weight (3100g) and length were both appropriate for gestational age. She had blue sclerae and also had multiple fractures of the left tibia on X-ray. The serum calcium and phosphorus and alkaline phosphatase were within normal limits. A diagnosis of osteogenesis imperfecta in a term female neonate was also made.
Both babies were managed conservatively with the orthopaedic surgeons and parents were counselled and taught how to handle the babies to minimise fractures. The babies were discharged on oral calcium and vitamin D supplements to the orthopaedic and paediatric outpatient clinics for follow up. They were however lost to follow up.
Figure 1
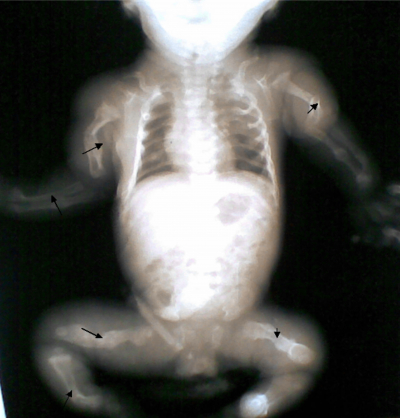
Figure 2

Discussion
The two babies presented in this report are possibly from new mutation because of the absence of a family history. We were however unable to do DNA sequencing or collagen culture for either the babies or the parents due to unavailability of these test in our facility. The previous history of two still births in the mother is also a possible pointer to the diagnosis as OI can also be a cause of still births; this and the presence of both healing and new fractures at birth, limb deformities and blue sclerae in the babies in this report suggest OI type II or III.
Available data on osteogenesis imperfecta in Black African children is limited; however some cases have been reported in African children including twins.9-12 Burundi,9 Nigeria10,11 and South Africa.12
Osteogenesis imperfecta was reported in monozygotic twins from Burundi who presented with multiple fractures especially of the femur when they started to walk and the diagnosis was confirmed by lower limb deformity, presence of wormian bones in the skull, blue sclera and tooth defects.9 Among the 10 pairs of twins reported by
A significant proportion of Nigerians with OI were also reported by Adeyokun to have type III disease.14 The 2 neonatal cases reported in Nigeria by Akinola et al were type II. These reports and the clinical presentation in this present report suggest that the severe forms of OI (types II and III) may be commoner in blacks.
Other manifestations include long bone abnormality (micromelia, rhizomelia), bowing of extremities, repeated fractures, easy bruising, teeth malformation (dentinogenesis imperfecta), short stature, macrocephaly with large frontanelles, triangular facies, low birth weight and length, small thorax causing respiratory insufficiency, vertebral abnormalities and compression e.g. kyphoscoliosis.
Important investigations include skeletal survey, collagen culture analysis which confirms the diagnosis, DNA sequencing, dual-energy x-ray absorptiometry scan (DEXA); however only the skeletal survey was done due to unavailability of these tests in our facility and the high cost of these tests in private centres (from where samples are sent abroad for analysis) in the country. Prenatal ultrasonography can also detect limb length abnormalities at 15 -18wks however there was no prenatal diagnosis in this case report.
There is no cure; but a multidisciplinary approach is required to manage the disorder. Early physical rehabilitation is encouraged, surgical procedures like intermedullary rod placement in bowed extremities, correction of vertebral abnormalities and relief of basilar compression may also be required.
Medical therapy involves use of intravenous bisphosphonates (e.g.pamidronate) reduces the incidence of fractures and increases bone density while reducing pain, however the intravenous route may limit its use.16 Two reviews also concluded that bisphosphonates increase bone mineral density but it is unclear whether they decrease fractures or reduce pain.17,18
Appropriate intake of calcium and vitamin D supplements is also essential. Both babies were only placed on Vitamin D and calcium supplements as no bisphosphonate was available. Other supportive measures include optimal caloric intake, audiological assessments and early introduction of hearing aids, physical therapy to improve joint mobility and achieve functional ability and psychological support for patients and their families. The complications include basilar impression, brainstem compression, recurrent pneumonia and respiratory insufficiency.
Prognosis depends on the type; types I and IV are compatible with full life span, type III is associated with reduced life span and patients with type II usually die within months to a year of life. The babies in the case report both fit into clinical type II disease (prenatal presentation, blue sclera) though definitive diagnosis using collagen culture could not be done and both babies were however lost to follow up.
Risk of transfer from a proband to an offspring is 50% and variability of expression may account for less or more severe disease in the child of affected parents. The risk of recurrence for an apparently unaffected couple is about 5 – 7% but this may be as high as 50% if one parent is a mosaic. Both parents in this report were apparently unaffected but we could not exclude mosaicism in them.
Conclusion
To our knowledge, this is the first report of this rare condition in twins in Nigeria and West Africa. The report also appears to support existing literature of the predominance of the more severe forms of the OI (types II and III) in black African children compared to type I in their Caucasian counterparts.
The finding of this pathology, especially in twins from apparently unaffected parents should remind the clinician of its existence and the various modes of inheritance. The importance of antenatal diagnosis should also be emphasized in order to guide the institution on prompt and comprehensive management of the baby and family. The establishment of at least one genetic centre in the country or a regional centre in West Africa which will be subsidized is recommended to help in diagnosis of genetic disorders such as OI to further improve management of these cases.